|
《临床生物化学》 > 第十九章 临床生物化学分析仪的性能与应用
第三节 临床生化自动分析的方法自动分析仪的应用决不仅仅是操作方法的变化,由于引进了一系列高精技术,所用的方法测定原理都有了迅速的发展。因此,用好自动分析仪的一个重要前提,必须对自动分析仪可以提供的测试方法类型、主要实验室参数和仪器室条件及仪器操作有所了解。
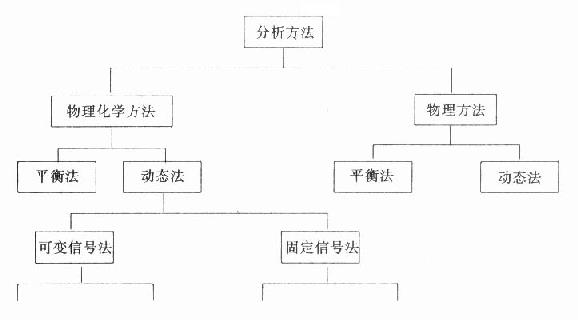
一、分析方法的分类临床生化常用方法根据其测定的原理可作如图19-5法分类。一类方法是物理方法,测定是物质固有的物理特性。另一类是物理化学方法,也就是将所测定物质进行一些化学转化后再进行测定。动态法是在所测定的物质浓度在不断变化中,由传感器获得相应的改变的信号进行计算的方法。平衡法是由传感器得到相对不变的信号进行计算的方法。现在越来越多被临床生化应用的酶试剂方法,无论是用自动分析仪记录或分光亮度计或普通比色计都包括在动态法的范围内。 (一)平衡法(终点法) 这类方法的特点是被测物质(酶反应的底物)在酶反应过程中应完全被转化或消耗掉,即达到反应的终点。通常这类方法操作简单,一般不需要作标准管,通过一些已知的理化常数,例如克分子吸光系数等不难将结果计算出来。其缺点是反应时间较长,特别不适合于离心式自动分析仪。还有少数酶反应,底物并未完全消耗掉,只是达到一个动态平衡,此时,往往需要同时作标准管。
图19-5 临床生化方法的分类 ⒈一步法 试剂酶的底物(S)就是所测的物质,在试剂酶作用于S后完全转化为产物(P),由于S和P具有完全不同的理化性质,从而可根据对S或P的浓度变化进行连续监测。使用最多的还是光度法,尤其是在340nm处测定NAD(P)H的生成或消耗量。在实际应用上最多的仍是和NAD(P)H有关的脱氢反应。如用乙醇脱氢酶测乙醇的含量或用乳酸脱氢酶测丙酮酸等。 ⒉酶偶联反应和测定酶活性方法一样,有时单用一个酶(辅助酶,Ea)不行,因为S和P之间往往无明显差异,不能直接测定,此时可以加入另外的酶(指示酶,Ei),将反应偶联起来,通过测定指示酶反应间接推算出所测物质,即第一个酶作用底物的浓度。 反应通式如下:
若Ei是一个脱氢酶,在反应过程中同时伴有NAD(P)H和NAD(P)间的转化,故不难在340nm处进行连续监测。另一常用的酶是过氧化物酶,可以通过新生态氧和一些色素原作用显色而进行测定。例如用已糖激酶(HK)法和葡萄糖氧化酶(GOD)法来测定葡萄糖等。 另外,还有一类少用的偶联酶反应,指示酶反应在辅助酶反应之前,而不是在后。
式中S1是欲测物质,往往另一底物S2不稳定,通过先行的指示酶产生S2,如乙酰CoA的测定。
其中草酰乙酸很不稳定,可通过苹果酸脱氢酶作用生成。 从理论上说酶催化反应都是可逆的反应,除水解酶外,大多数酶往往都不易将底物完全转换或消耗掉。常可通过增加不需测定的另一底物浓度,改变pH,使用捕获剂(trap-ping agent)等改变平衡点,使反应偏向一侧,达到或接近反应完全。 (二)动态法 动态法在自动分析仪中的应用十分广泛,但是动态法的分类一直比较混乱。一般可分为可变信号法和固定信号法两类。 ⒈可变信号法 ⑴直接法即根据所得到的吸光度变化直接计算结果:由于对所收集数据多少有很大意义,所以又区分为一点法、二点法、多点法。一点法是在一个预先设置时间测定各个标本吸光度。二点法则是在一点法基础上多测一点,即在酶反应仍未进行时的吸光度,和一点法相比可以通过空白测定扣除这部分误差,但是仍无法了解反应过程,避免不了延缓期或非线性反应所引起的误差。多点法如使用确当,对标本空白和反应过程都有详细了解,可用以下两法: 一法是求出不同测定点间吸光度的差异即所谓delta法。各点吸光度差异可用б=△A=An-An-1计算出。根据这些数据可以观察反应是否符合线性,并计算出被测物质浓度。偏离线性的数据在计算结果时应摒弃。很多早期自动生化仪都使用此法。另一法则是使用回归法,根据一定公式如线性公式y=a+bx对获得的多个数据进行计算,求出斜率b和截距a。这类方法也可用于浓度计算。和delta方法相比,不仅可适用于线性反应,也可适用于其它非线性反应。 ⑵导数法在此法中使用电子线路计算出反应瞬间的一阶和二阶导数:bekman system TR就使用了这样方法,它可以同时计算出一阶和二阶导数,用二阶导数来观察哪一段反应为线性反应,并用线性反应段上的一阶导数计算出物质浓度。此法单用导数方法并不记录吸光度变化(△A),所以此法所提供的信息往往少于回归法,例如无法观察到反应中底物的消耗情况。 ⑶积分法此法原理是运算放大器对反应曲线二个节段的吸光度进行积分。并计算出部分面积之差。Vitatron分析仪用此差值来考虑线性段,但仍用△A法计算结果。 ⒉固定信号法测定原理是随着反应进行,通过不断添加试剂使吸光度变化在很窄范围内,此时所测的是加试剂的量,并依此来计算物质浓度。最典型例子是以对硝基酚作为指示剂的乙酰胆碱酯酶方法,在反应过程中不断加入碱以中和酶水解产物乙酸以维持pH不变,然后根据所加碱量计算酶含量。这一类方法由于操作费时麻烦,目前应用很少,但不应忘记这类方法能维持反应条件如pH的恒定性,其它如在NADH参与的反应中不断加入NADH有助于避免底物的消耗,在一些特殊情况下可能还是有用的。 由此可见,假如从方便简单不需复杂仪器而言,则一点和二点法应为首选。而回归法、积分和导数法不易进行。如从可靠性来进行比较,则多点回归法应居榜首,一点和二点法最不准确。 回归法的缺点是此法可以不考虑收集的数据是否可靠,不管各数据是否偏离曲线或明显不呈线性,也可求得一个线性方程式。所以,一个好的回归方程式还应给出标准差和有关参数,这样即可用来评价用回归法所得结果的可靠性。根据Pardue意见,使用较多数据且设计良好的回归方法是首选的。 (三)固定时间法(二点法) 固定时间法在自动分析仪中的应用,有助于我们解决反应的特异性问题,最明显的例子就是苦味酸法测肌酐和溴甲酚绿法测白蛋白。人们早就知道很多物质如维生素C、乙酰醋酸、葡萄糖、果糖以及某些药物也能和苦味酸呈色。近年来发现这些干扰物质呈色较慢,提出用自动生化仪只测定开始一分钟的反应,明显提高了苦味酸法测肌酐的特异性。用溴甲酚绿测白蛋白法,此法由于简单易行,在70年代取代了经典的盐析法,但随即发现一部分球蛋白如α球蛋白也能和溴甲酚绿结合,但这类反应比较迟,所以目前更多地使用自动分析仪测定开始一分钟的反应来计算白蛋白量。 固定时间法之所以受到愈来愈多注意,更重要因素是酶试剂的应用(详见有关章节)。 (四)空白对照方法 由于使用了各种高新技术,在自动生化分析仪中人们可以根据实际情况,使用各种空白和对照方法。 ⒈固定法空白①试剂;②标本+盐水;③标本+空白试剂;④标本+灭能试剂;⑤标本+完全试剂(时间不同); ⒉动态法空白①试剂空白;②标本+空白试剂。 从理论上说固定空白法用“标本+完全试剂”是最好的办法,此法可以扣除各种因素的误差,如试剂、比色杯等,这也是在自动生化仪应用较多的方法,而用手工法是不易做到的。动态空白仅用于少数试剂不稳定方法,如以固红B测天门冬氨酸转氨酶。
二、主要实验参数的选择(设置)一般自动分析仪有12个主要数据,即波长、温度、标本及试剂量、空白时间(Tb)、开始收集实验数据时间(Ti)、实验数据收集窗时间(TW)、实验数据收集完成时间(Td)和最后时间(Tf)、速率时间(Tr)、线性限度(L)、异常吸收率(ABN)、曲线拟合、浓度因素(F)。这些参数都是通过方法学的研究之后,根据反应的具体情况加以确定的。 (一)波长 根据分光亮度计的光吸收曲线或者一个比色杯不同波长的反应曲线选择分析波长和空白波长。对快速反应,以光吸收曲线的低凹区波长为空白波长,光吸收曲线中吸亮度最大而较平坦区的波长为分析波长。 (二)温度 一般均选用30℃。 (三)样品量及试剂量 可根据手工法按比例缩减或者重新设计。要考虑到检测灵敏度、线性范围,尽可能将样品稀释倍数大些,以降低样品中其它成分的影响。 (四)分析时间的确定 用高、中、低浓度的标准液进行实验用研究模式,进行酶促反应时间曲线或反应速度时间曲线的观察,根据吸收率动态变化的具体数据,确定有关的分析时间。 ⒈Tb 终点法,吸光度上升型,一般选择反应尚示开始而试剂和样品已充分混合均匀,一般定为4秒。动态法的Tb=Ti。 ⒉Ti 终点法选择反应接近平衡期,动态法则选择接近进入线性期。这个参数不很严格。如果Ti定的太早,过多的不符合要求的数据点将舍弃。Ti定的太晚,数据收集时间就要延长,在动态法还可能使底物耗尽的发生机会增多。在终点法Ti大致定于Tf的70%或接近于反应完全时,动态法确定Ti时,主要考虑避开酶反应的延迟期以及试剂和样品混合后温度不平衡期。 ⒊TW 根据高、中、低浓度标准液反应速度时间曲线加以确定。要求各标准液的吸光度变化在该时间内均在线性判断标准范围内。终点法一般为4-10秒。在动态法中,为了使反应不出现未反应(NR)的标志,在Tb与Tf之间至少有8mA的吸收率变化。在快速反应时,TW从30秒开始,按10秒向上递增。在慢反应可达120秒。TW不应太长,否则可以偏离线性,也减低工作效率。 ⒋Tr Tr值的大小对方法的精密度有较显著的影响。为了保证方法的精密度,使△A>1.0mA是必要的。如Tr时间短,△A太小,测定方法的精密度就降低。一般情况下,Tr定为20-60秒。在现有程序中,是以正常低值标本的△A>1mA来决定Tr值。如ALT测定,正常低值为7u/L,Tr=7u/L/2.57=2.7mA/60秒,30秒=1.35mA,故选30秒为Tr时间。 (五)线性判断标准(线性限L) 指在数据收集窗时间内吸收率变化的允许范围。作为终点法,从理论上讲,化学反应达到平衡(终点),吸光度应该稳定不变,也就是说,在TW内吸收率变化应该是零,即线性判断标准应该定为零。在实际中,由于信号有一定的飘移,更主要是测定要求快速,是在反应还没真正到达终点时进行测定,加上自动分析仪检测的灵敏度大多较高,精度可达到0.1mA,所以在TW期间的吸收率会有一定的波动。不同的化学反应或酶促反应,吸光度变化程度不一。要根据实验结果选择一个适当的吸光度变化范围作为线性判断标准,以此来判定反应是否达到了平衡。这个标准值如果定的太小,反应也很难达到要求的标准,出现非线性的机会太多,或者要延长观测时间,降低工作效率,这就不符合快速分析的要求。相反,这个线性判断标准定的太大,就失去了判断线性的意义,出现反应根本没有达到终点就错误地判为达到标准,影响测定结果的可靠性。在终点法中,L单位是mA/2秒,大多选用1.0mA/2秒。如出现反应不完全,可按0.5mA顺序增加。在动态法中,L单位是mA/秒,一般从0.1mA/秒开始,如果试验结果出现非线性(NL)情况多,可以增大,按0.01mA递增,但不可超过0.1mA。 (六)异常吸收率限度 这一参数主要根据每个方法实验测定的线性范围来确定。即用吸光率作为纵轴,标准液浓度为横轴,绘制标准曲线。这个线性段向非线性段转折拐点的相应吸收率值即为异常吸收率限度。终点法吸收率超过此值被标志为超出限度(XL)。在动态法中,试剂空白的吸收率超过ABN,ABN被标志为试剂吸收率异常(AB),测定杯的吸收率超过ABN时,ABN被标志为底物耗尽(EX)。
三、仪器室条件及仪器操作(一)仪器室条件 ⒈接入仪器室的电源线应大于仪器及室内电器所指定的安培容量。 ⒉电压不稳定或低于额定电压的地区需加装大于指定功率的电压稳压器。 ⒊经常停电地区可装大于指定功率的不间断电源,电源的工作时间任选。 ⒋应安装空调器以保持恒温与无尘。空调器的制冷量应大于仪器的产热量,室温波动每分钟应不大于1.33℃。试验温度对室温是有要求的,各型号仪器要求不一,一般30℃试验的室温是18-25℃,37℃试验的室温要求是18-30℃。 (二)仪器操作 各种型号的仪器都有规定的日常操作程序,应按照说明书要求进行。
…… 第三节 分子生物学实验诊断技术 第四节 诊断分子生物学技术的临床应用 第十九章 临床生物化学分析仪的性能与应用 第一节 临床生化自动分析仪的类型 第二节 临床生化自动分析仪的性能评价与合理选用 第三节 临床生化自动分析的方法(当前页) 第二十章 临床生物化学方法的选择、建立和评价 第一节 临床生化方法的选择 第二节 临床生化方法的建立 第三节 临床生化方法学的评价 …… |