|
《实用免疫细胞与核酸》 > 第二十一章 原位杂交技术在染色体和电镜水平的应用
第一节 原位杂交技术在染色体铺片的应用近年,不少科技工作者将原位杂交组织细胞化学技术(ISHH)应用于细胞分裂中期(Metaphase)和分裂间期(Interphase)的染色体铺片上,利用标记的核苷酸探针与之进行杂交,可在光镜或电镜水平检测到不同的特异性的标记物。目前,原位杂交组织化学在染色体铺片的应用主要在三个方面:染色体的基因分配或基因图(Gene assignment)的研究、癌细胞或癌前组织的染色体异常的间期细胞遗传学分析研究和性染色体对胚胎的生前诊断。在依次介绍这些方法的应用以前,我们先简要介绍一个有关染色体的基本的知识和染色体铺片的制作方法。
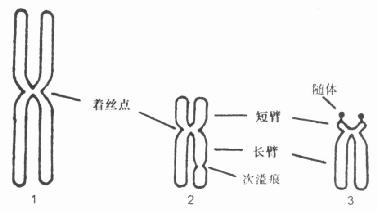
一、有关染色体的基本知识及制片(一)染色体的形态结构体细胞的染色体是46个,23个,其中22对是常染色体,一对是性染色体。男性一对XY,女性为XX。染色体的形态随着细胞周期的不同而有所改变,在光学显微镜下所看到的染色体是细胞分裂中期染色体(metaphase chromsome)。 每个染色体含有两条染色单体,呈赤道状彼此分离,只有着丝粒处相连。根据着丝粒的位置分为三种类型,中部着丝粒型,亚中部着丝粒和端着丝粒型(图21-1)。
图21-1 正常人体细胞的三种染色体 1.中部着丝点染色体;2.近中部着丝点染色体;3.近端部着丝点染色体 1.非显带染色体特征分为七组 A组(1~3):为最大的具中部着丝粒染色体,这组染色体相互间很易区别。第1号和第2号染色体大小相似,唯第2号染色体为近中部着丝粒染色法。第3号染色体较1、2号染色体小,为中部着丝粒染色体。 B组(4~5):为大的具中部着丝粒染色体。2对染色体之间在形态和长度上较难区别。 C组(6~12号和X):为中等大小的具中部或近中部着丝粒染色体。这组染色体较难区分,其中第6、7、11号和X染色体为中部着丝粒染色体,第8、9、10和12号染色体为近中部着丝粒染色体。女性为2个X染色体。男性只有1个X染色体。 D组(13~15号):为中等大小的具近端着丝粒染色体。在其短臂上有随体。与他组染色体有明显区别。但3对染色体之间较难区别。 E组(16~18号):为小的具中部或近中部着丝粒染色体。第16号染色体为中部着丝粒染色体,第17号和18号染色体为近中部着丝粒染色体。不过,着丝粒位置第18号较第17号染色体更近端部。 F组(19~20号):为更小的中部着丝粒染色体。2对染色体之间,形态上很难区别。 G组(21~22号和Y):为最小的近端着丝粒染色体。第21号和22号染色体大小相似,且短臂上常连有随体。Y染色体常比第21和22号染色体大、染色深。且无随体。Y染色体长臂2个染色单体比较靠拢,长臂末端也较模糊。 2.G带染色体的特征 第1号染色体:识别并不困难,但初学者易把长短臂颠倒。短臂的近侧1/2处有2个深带。远侧有一半几乎为浅染区;短臂共分3区。界标为近侧的深带(2区1带)和中段的深带(3区1带)。长臂近侧为一窄的浅带。中段和远侧段各有2条深带;长臂共分4区,界标为次缢痕远侧的浅带(2区1带)、中段的深带(3区1带)和远侧的深带(4区1带)。 第2号染色体:短臂可见4条深带。中段的2条深带常互相靠近,表现为1条深带;短臂共分2区,界标为1浅带(2区1带)。长臂可见4~7带,近侧着色甚浅,中段及远侧着色较深;长臂共分3区,界标为2个浅带(2区1带和3区1带)。 第3号染色体:短臂近侧可见2条深带;远侧可见3条深带,其中远端的1条带较窄,染色浅;短臂分为2区,界标为1浅带(2区1带),长臂近侧和远侧各有1条较宽的深带:好的标本近侧深带可分为2条深带,远侧深带可分为3~4条深带;长臂分为2区,界标为1浅带(2区1带)。长臂深带宽度略大于短臂深带。 第4号染色体:短臂有1~2条深带,只1区。长臂有均匀分布的4条深带;长臂分3区;界标为2浅带(2区1带和3区1带)。 第5号染色体:短臂有1条深带,只1区。长臂有5条深带,中段的3条深带常靠近;长臂分3区,界标有1深带(2区1带)和1浅带(3区1带)。 第6号染色体:短臂近侧有1宽的浅带,远侧为1深带;短臂分2区,界标为1浅带(2区1带)。 第7号染色体:短臂有2~3条深带,近末端带着色很深;短臂分2区,界标为1深带(2区1带)长臂有3条深带,近侧和中部的2条带较深;长臂分3区,界标为2深带(2区1带和3区1带)。 第8号染色体:短臂有2条深带,远侧深带较近侧深带着色深;短臂分2区,界标为1浅带(2区1带)。长臂有3~4条深带,远中的深带着色较深;长臂分2区,界标为1深带(2区1带)。 第9号染色体:短臂有1~2条深带;短臂分2区,界标为1深带(2区1带)。长臂2条深带。长臂分3区,界标为2深带(2区1带和3区1带)。 第10号染色体:短臂有2条深带,近侧较近侧深带着色浅;短臂只1区。长臂有3条深带,近侧深带着色更深;长臂分2区,界标为1深带(2区1带)。 第11号染色体:短臂有1~2条深带;短臂只1区。长臂有2条深带,在近侧深带与着丝粒之间有1宽的浅带;长臂为2区,界标为1浅带(2区1带)。 第12号染色体:短臂有1条深带;短臂只1区。长臂有3条深带,中间的1条深带宽,近侧深带与着丝粒的距离较第11号染色体近;长臂分2区,界标为1深带(2区1带)。 第13号染色体:长臂有4条深带,中间2条深带宽;长臂分3区,界标为中间的2条深带(2区1带及3区1带)。 第14号染色体:长臂有45条深带,近侧的第2条带和远侧的带着色更深;长臂分3区,界标为2条深带(2区1带和3区1带)。 第15号染色体:长臂有4条深带,近侧的第2条带较宽;长臂分2区,界标为1深带(2区1带)。 第16号染色体:短臂有1~2条深带,短臂只1区。长臂有2条深带,近中部深带明显;长臂分2区,界标为1深带(2区1带)。 第17号染色体:短臂有1深带;只1区。长臂1~2深带,远侧深带较宽,在深带与着丝粒之间有1宽的浅带;分2区,界标为浅带(2区1带)。 第18号染色体:短臂为1浅带区。长臂有2条深带;为2区,界标是1浅带(2区1带)。 第19号染色体:着色最浅。着丝粒周围为深带。短臂和长臂均为1区。 第20号染色体:短臂有1深带,较宽;短臂和长臂均为1区。长臂有1深带,较宽。 第21号染色体:最小的1个染色体。长臂紧按着丝粒处有1深带,较宽;长臂为2区,界标为1深带(2区1带)。 第22号染色体:较21号色体长,长臂紧接着丝粒处有1深带,较窄。短臂和长臂均为1区。 X染色体:短臂中部有1条深带;分2区,界标是深带(2区1带)。长臂有4条深带,其中近槽深带最宽;分2区,界标是近侧深带(2区1带)。 Y染色体:长臂远侧半色着深,为1深带;短臂和长臂均为1区。 现在国内流行一种辨认G带每个染色体的口诀,稍加修改如下: 一秃二蛇三蝶飘,四象鞭炮五黑腰; 六号短空小白脸,七上八下九苗条; 十号长臂近带好,十一低来十二高; 十三十四十五号,三个长臂一二一; 十六长臂缢痕大,十七长臂带脚镣; 十八人小肚皮大,十九中间一黑腰; 二十头重脚脚底轻,二十一带宽身体小; 二十二带小身体大,Y长一宽近末端; X短臂一、长臂三,中间象个工字般。 (二)染色体G显带技术1.原理染色体的化学成份是核酸和蛋白质两部分。核酸以DNA为主,蛋白质有组蛋白和非组蛋白两种。因此染色体经蛋白水解酶类物质处理后,蛋白质已被水解而使DNA分子中碱基暴露,由于碱基中G/C和A/T组合的比例不同,对染料结合的程度不一,如这一段A/T碱基的成份多,则Giemsa染料易与它结合成深染;另一段G/C碱基的成份多,则Giemsa染料不易与其结合,结果成淡染,由于整条染色体上A/T和G/C的分布不匀,这样在染色体上呈现出深浅不一的条纹或者称带纹,该技术用Giemsa染色,故其带型称为G带。 2.操作过程 (1)按常规染色体技术制片。 (2)制好的染色体玻片,在80℃烘箱中,烘烤2h,置于室温。 (3)4℃冰箱PBS液5min。 (4)4℃冰箱PBS缓冲液含胰蛋白酶终浓度0.025%,消化10min。 (5)蒸馏水洗净胰酶。 (6)Giemsa染色20min。 (7)蒸馏水洗净染色液,晾干镜检。 4℃低温胰酶处理片子,其优点是:时间较长,易掌握。配制1次试剂可用1~2个月。 (三)高分辨染色体G带技术1.原理常规的G带显带技术,在人类染色体中仅观察到320~450条带纹,对于一些染色体细微结构异常的识别是不够的。 氨甲碟呤抑制二氢叶酸还原酶,阻止叶酸转变为四氢叶酸,从而干扰了脱氧胸腺嘧啶核苷酸的合成,阻止了DNA的复制,使细胞被阻止在S期内。胸腺嘧啶核苷解除氨甲喋呤的作用,让被阻滞的细胞同时开始进行DNA复制,进而同步分裂;放线菌素D可增加染色体的长度,能显示更多的亚带。放线菌素D与核酸相互作用:①插入脱氧鸟嘌呤核苷酸和脱氧胞啶核苷酸之间,使DNA变长;②能与染色体缩短有关的特殊蛋白和DNA结合,因此,抑制了染色体正常的收缩过程。秋水仙胺抑制纺垂丝的形成,可以得到较多的晚前期、前中期、早中期的分裂相。这样染色体带纹可增加到500和850条,甚至可达1200~2000条之多。这一步对于研究较细小的染色体缺陷的基因定位,具有很大的意义。 2.材料和方法 (1)4ml培养液(为从日本进口的RPMi 1640或从美国进口的F10液)虽自制的小牛血清1ml。每ml加青、链霉素各100单位,pH=7.2。 (2)每瓶培养液内,另加自制的PHA或重庆产的PHA2.5mg。 (3)取外周血2ml,分别装入含培养液的4个培养瓶内。 (4)37℃孵育箱内培养72h。 (5)加入氨甲喋呤,最终浓度10-7mol/L,继续培养17h。 (6)用不含血清的培养液冲洗两次后,将上清液吸出,再加入含胸腺嘧啶核苷(最终浓度为10-5mol/L)的培养液,继续培养3h。 (7)再加入放线菌素D,最终浓度为3μg/ml,培养2h。 (8)加秋水仙胺,最终浓度为0.03μg/ml,培养2h。 (9)按常规染色体技术制片。 (10)按一般常用的G带显带技术,显示带纹。 3.注意事项 (1)高分辨染色体,重叠多,应增加固定次数和固定时间,可置于4℃冰箱存放24h后制片,用高距离滴片,以使染色体分散开。 (2)细胞培养液同步化后,如果用5-溴-2脱氧尿核苷(5-bromodeoxyuridine,BrdU),则需加黑物包装进行暗培养。 (四)外周血培养及染色体制片技术1.国内应用的外周血培养染色体技术 (1)原理:外周血染色体制备是目前应用最广泛的细胞遗传学诊断技术,亦是基本的实验技术,由于取材容易,培养过程比较简单,短期内可以得到结果,所以其实用价值很高。 血液中含有红、白两类细胞,它们均是处于未分裂的间期细胞。红细胞没有核,无分裂能力;白细胞类虽有细胞核存在,但是外周血中处于休止期。植物血球凝集素(简称PHA)能促使淋巴细胞和单核细胞转化为具有分裂作用的母细胞。染色体只有在细胞分裂中期时最典型。秋水仙素类药的药物能抑制纺锤丝的形成,使细胞分裂停止于中期,低渗处理使细胞膨胀,细胞膜破裂,染色体分散,这样就可便于分析。 简意流程图:
(2)操作过程 ①采血:无菌干针筒从静脉取血1~2ml,用肝素抗凝(约每毫升全血内含肝素100单位)。 ②培养液配制:RPMI 1640 4ml 小牛血清 1ml 1%PHA(自制)0.2ml 调节 pH为7.4后置于-20℃冰箱 ③标本接种:每5ml培养液加入抗凝全血0.5ml。 ④培养细胞:将接种好的培养瓶置于37℃恒温箱中培养72h。 ⑤阻止分裂:培养至68h,加秋水仙素,最终浓度0.02μl/ml,摇匀后置于37℃恒温箱中继续培养4h。 ⑥收获:培养物混合后,置于10ml离心管内,离心10min(1000转/min),去上清液。留下培养物。 ⑦低渗处理:低渗液可用0.56%的KCl(或0.075mol/l KCl)5~7ml,用吸管打散沉淀物,置于37℃水浴箱中保温20min,然后加入固定液(3份甲醇:1份冰乙酸)1ml用吸管打匀,预固定1~2min。 ⑧离心:1000转/min离心10min。 ⑨固定:吸去上清液,留下沉淀加上述固定液6~8ml,用吸管将沉淀物打散,固定30min后离心,1000转/min离心10min,取出吸去上清液留下沉淀。 ⑩重复上述固定离心1次。 ⑾标本制作:最后1次固定、离心后吸去上清液,留下沉淀再加少量新鲜固定液约0.3ml打散细胞悬浮液即可进行制片。制片之前先将载玻片经清洁液洗净处理后,置于冰箱内制成冰片。取冰片1张滴上2~3滴细胞悬液。利用冰片的表面张力关系使液体迅速向玻片四周散开,与此同时用嘴轻轻吹向滴片处,以助其更快散开,然后在酒精灯火上通过7~8次后,自然干燥或烤干均可。 ⑿染色:Giemsa染液1份和pH7.4磷酸缓冲液9份,混合后,染色20min,蒸馏水洗去余下染液,晾干,镜检观察。 (3)注意事项 ①无菌操作是关键。培养液不得污染。 ②所用药品不得失效,特别是PHA,既要使淋巴细胞转化,又不能过量。 ③小牛血清优质、无菌。 ④秋水仙清素低浓度4~6h效果最佳,过量则染色体易收缩。 ⑤固定液和Giemsa染液要求新鲜配用。 ⑥培养时间72h较好。 (4)各种物品清洗与消毒 ①玻璃器皿用后,立即用自来水冲洗,再用洗衣粉刷洗,然后用自来水冲洗。待干,泡入清洁液24~48h,取出,自来水冲洗,再用双蒸水冲洗,干后备用。 ②接触洗液时,带上橡皮手套,围裙等防护品。 常用清洁液配制: 重铬酸钾25g 水200ml 浓硫酸1000ml ③先将重铬酸钾在水中溶解,然后慢慢加入浓硫酸,边加边搅拌,防止过度发热。使用3~6月后检查,若变绿表示失效。 ④G5、G6漏斗的处理:水洗净、晾干,于浓硫酸泡(或洗液中泡)24h,再用蒸馏水冲洗至加入1%BaCl2滴无白色沉淀为止。包装消毒,15磅20min。 (5)橡皮塞的处理 新的橡皮塞洗后,先用0.5N NaOH煮沸15min,再用0.5n HCl煮沸15min,流水冲洗10min,用双蒸水泡24h,双蒸水冲洗,晾干,15磅20min高压灭菌。 (6)金属器械清洗 先用纱布或纸擦去防锈油,然后用洗涤液清洗,再用清水或蒸馏水洗净,擦干或烘干备用。 2.外周血细胞培养法(Bhatt B and McGee JOD,1990) (1)收集静脉血(无菌),加肝素抗凝(20单位/ml); (2)加0.8ml全血入每个培养瓶(含10ml培养液),37℃培养72h; 培养液配制:RPMI 76ml(Gibco,Cat.No.041-1875M) 小牛血清 20ml PHA 2.0ml (3)加100μl Brdu于培养瓶中,37℃再孵育16~17h; (4)离心(1200rpm)8min,弃去上清液,沉淀中加入10ml RPMI 1640培养液,混匀; (5)重复步骤(4); (6)沉淀中加入10ml新配制的完全培养液(含2.5μg/ml胸腺嘧啶),重新置于37℃孵育6~7h; (7)在收获培养细胞前15~30min,可加Colcemid,但当同时应用BrdU时这就并非十分必要。因为BrdU是胸腺嘧啶核苷的类似物,能使染色体在富含异染色质处断裂分解,从而使显带明显; (8)1200rpm离心培养细胞,弃去上清液,加入1ml 0.56% KCl(事先预热至37℃),继续加入0.56%KCl使总容量达到10ml,在37℃孵育10min; (9)如上述离心,弃去上清液,在沉淀的细胞内加入新鲜的冷固定剂,(甲醇:冰醋酸=3:1,新鲜配制,保存在冰上),置冰上至少20min; (10)重复步骤(9)3~4次,直至细胞悬液清洁无色; (11)可较长期保存在冰的固定剂内(-20℃)或再离心后加入0.5~1.0ml新鲜固定剂,置于冰上; (12)玻璃载片放在Decon(清洁液)或其它的清洁液内过夜,次日用自来水冲洗,继之蒸馏水漂洗,在60~75℃干燥,然后把载片孵育在2%丙酮液内,室温,60min。自来水冲洗,再在60~75℃干燥; (13)每张载片上加10μl的细胞混悬液,空气干燥,在24h后可应用于杂交实验,也可保存在-4℃达8周之久(试剂配制法见附录四)。
二、应用ISHH技术对染色体制片、基因分配(Chromosomalassignment of genes)的研究(一)基本原理 染色体上基因位置分配的研究,又叫染色体图的研究,包括基因名称、基因在染色体的位置及与相邻基因的距离及其核酸序列的研究。研究基因图的方法包括家系的连锁分析法、体细胞杂交法、重组DNA技术和原位杂交法。果蝇的线形基因图和大肠杆菌、T4噬菌体两者的环状基因图都已绘成,目前正致力于人类基因图的绘制。人类基因约有5万~10万个,根据第八次国际人类基因定位工作会议,已定位的人类基因总数达1479个。 ISHH技术对染色体基因图的研究,是用同位素或生物素、地高辛标记的核酸探针,直接同中期的染色体铺片行杂交。早期的研究是用于重复序列DNA的定位,如编码核糖体的基因18S和28S核糖体RNA定位于第13~15号染色体和第21~22号染色体短臂次缢痕区。5S核糖体RNA定位于第1号染色体上长臂的远侧Iq42~Iq43。近年已能进行单拷贝基因的定位,如胰岛素基因定位于IIp 15上、C-mos原癌基因定位于Sq22上、N-ras原癌基因定位于IPI34。在染色体11的短臂上,利用ISHH技术证明4个基因依次排列,从短臂的远端依次为胰岛素、β-球蛋白、HRASI和PTH。 (二)操作步骤(Bhatt and MCGee, 1990) 在前面已介绍了制备血培养染色体铺片的方法。下面介绍的是在染色体制片上用免疫金银染色法进行原位杂交的基因定位显示、结合Giemsa染色显带技术的操作步骤。 1.移除内源性RNA (1)加100μl RNA酶溶液,覆以盖玻片,放在有少许2×SSC溶液的湿盒内孵育,37℃,60min。 (2)在2×SSC溶液内移除盖玻片,以2×SSC洗2×3min。 (3)等级酒精系列脱水,10%,50%,70%,90%和100%,各1min,各1min。空气干燥。 2.探针标记和纯化用缺口平移法将生物素11-dUTP标记在DNA核酸探针上。 3.变性与杂交 加20~100ng生物素标记探针到杂交液中,加入5μl 20mg/ml人或鲱鱼精子DNA,总容量为30~40μl,离心5s,混匀后,滴于染色体铺片,覆以盖玻片,盖片四周以橡皮泥封固。放入盛2×SSC溶液温盒内75℃7~8min,使之变性,继之于37℃孵育16h。 4.杂交后漂洗2×SSC溶液内移除盖玻片,将载片浸入含50%甲酰胺的2×SSC溶液内,pH7.2,20min,42℃。然后作下列程序漂洗 溶液 漂洗时间温度 2×SSc 20min42℃ 1×SSC20min 室温 0.1×SSc 20min 室温 5.免疫细胞化学显示 (1)孵育载片于PBt 15min,将片缘及背面擦干,加100μl一抗于载片(兔抗生物素抗血清1:100,以PBT稀释),孵育于37℃40~60min。 (2)快速用PBT冲洗,擦干片缘及背面,加100μl二抗抗血清(羊抗兔IgG结合10~20nm金粒,以PBT1:50稀释)于载片染色体铺片部位,37℃孵育45~60min。 (3)PBS洗3×5min。 (4)蒸馏水洗3×5min。 (5)加数滴银溶液于载片上,在光镜下监测,大约需5~18min可见在染色体上出现银斑点。 (6)蒸馏水洗。PBT配制见附录3。 6.显带复制(Replication banding) (1)应用5μg/ml Hoechst 33258在2×SSC溶液内,暗室中染载片20min。 (2)蒸馏水洗,以2×SSC封固,覆以盖玻片,盖片四周封以橡皮泥。 (3)紫外光照射1~16h。 (4)蒸馏水洗和以10%Giemsa(磷酸缓冲液稀释,pH6.8~7.2)染色10min。 (5)蒸馏水冲洗,空气干燥,以DPX封固。 (6)光镜下观察,可见棕色的银斑点在染色体上的定位,并显示其与染色体分带的关系。
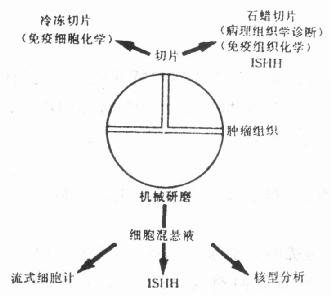
三、利用ISHH技术对肿瘤或癌前组织的细胞间期染色体铺片进行研究(一)基本原理 在恶性的或癌前病变的组织,染色体组含量与结构的畸变在部分病例中与疾病的预后相一致。由于染色体基因的变化可能导致肿瘤的发生与进展,利用一些技术预测这些遗传物质的变化可为恶性肿瘤的检测及了解其进展和预后提供资料。流式细胞计(FCM)和染色体组型,即核型分析(karyothping analyses)等技术曾被用于这个领域的研究。FCM可测定肿瘤细胞DNA的含量,但它不能显示特异性染色体的结构异常,而且对微量的DNA含量改变难以检测。核型分析可以显示染色体结构和含量的变化,但要分析肿瘤细胞的核型只有在细胞培养以后。细胞培养时细胞的高度分裂指数和细胞的生长会导致染色体物质的丢失,而且核型分析不能显示特异性DNA探针的原位杂交技术,能够检测在细胞分裂间期细胞核内染色体数量和结构的异常变化。因此,这项技术又被称为“间期细胞遗传学分析法(Interphase Cytogenetics Analysis, ICA)。间期细胞遗传学的基本原理是利用合成的特异性DNA探针,标记以同位素、生物素、地高辛或荧光素,对外周血、肿瘤细胞或癌前组织的离散培养细胞的染色体铺片或切片进行DNA-DNA原位杂交,其杂交定位以荧光法、放射自显影或免疫组化法显示。目前ICA已广泛应用于生前诊断、肿瘤组织活检材料的诊断和基因异常的诊断。在肿瘤细胞方面,在乳腺癌,神经系统肿瘤、睾丸肿瘤,妇科肿瘤和膀胱肿瘤等均有实验报告。现已能提供的DNA探针有染色体1,7,8,9,10,15,16,17,18和性染色体X、Y,以及识别一些染色体特征性部位如着丝点(centromeric region)、染色体端极区(telomere region)的特异性重复靶核苷序列和随体的DNA探针,多数为合成的寡核苷酸探针,在应用上采取ISHH和FCM以及免疫组化相结合(有时还结合核型分析)的综合研究法(图21-2),也可用多种标记物显示不同颜色或不同标记在一张切片上同时显示多个染色体的结构或DNA含量异常,如异硫氰酸荧光素(FITC)和罗丹明200结合,前者显示黄绿色,后者显示红色,ABC-DAB显示法与地高辛-碱性磷酸酶系统相结合,前者反应物为棕色,后者为紫蓝色。这种联合应用法又叫多靶点原位杂交法(multiple-target ISH)。
图21-2 肿瘤组织处理流程图 (二)基本操作方法 1.玻片与组织前处理 (1)肿瘤细胞混悬液的制备:新鲜的从活检或外科手术或尸检获得的材料建议按图21-2的流程处理进行综合研究。用以制作细胞混悬液的组织在处理前可保存于液氮内。组织块在玻皿中切碎或用细胞打碎机打碎,用100μm孔直径的尼龙网过滤器滤过,固定在70%乙醇,-20℃,如保存于-39℃可保存数月至数年。 (2)分离的细胞由于表面有细胞质的覆盖,在荧光显微镜下会显示强烈的自动荧光,从而掩盖了ISHH的信号。可采取下列两种方法移除覆盖的细胞质; ①乙醇固定后的细胞,应用以前在新鲜制备的甲醇/醋酸(3:1)固定液内0℃,5min。滴2~3滴固定后的细胞混悬液于涂有粘附剂的载片上,空气干燥,然后快速浸入70%醋酸内10~60s。应用蒸馏水洗,在100%乙醇内脱水,空气干燥。 ②应用胃蛋白酶蛋白水解作用。加5μl细胞混悬液于载片上,空气干燥30min或在80℃烤箱中干燥。胃蛋白酶(2500~3500单位/mg蛋白质,Sigma,USA)应用0.01mol/L HCl稀释至50~400μg/ml,10min 37℃;再用0.03% H2O2和PBS漂洗,以4%多聚甲醛固定,0℃,5min;载片脱水,空气干燥,在80℃加热变性30min。这个方法较①更好,能移除蛋白质、暴露核DNA和有助于探针的穿透,最重要的能较好的保持形态学的结构,以利于ISHH的荧光信号的显示。应用①法,细胞经常呈扁盘状,而且细胞质的自动荧光未完全消失,影响ISHH荧光信号的显示,但实验表明,必须严格掌握胃蛋白酶的浓度,过低,由于细胞质的覆盖,基因拷贝数会减低,而过高浓度的胃蛋白酶会导致过度消化影响细胞的形态结构。 (3)组织切片处理 ①冷冻切片:切片厚5μm,放置有粘附剂的载片上,空气干燥,固定在4%多聚甲醛-PBS溶液,0℃,15min。固定后以PBS和0.01n HCl漂洗,以胃蛋白酶(20~100μg/ml,用0.01N HCl配制),37℃,30min,PBS漂洗2×5min,在4%多聚甲醛内固定15min(0℃),PBS洗2×5min,系列等级乙醇脱水,空气干燥。杂交前,在80℃加热30min使之变性。 ②石蜡切片:切片厚5μm,漂洗在40℃温水中,捞片于有粘附剂的载片上,空气干燥,56℃加热1~16h,二甲苯脱蜡2×3min甲醇冲洗2×5min。应用1%H2O2(用甲醇配)溶液封闭内源性过氧化物酶活性。以甲醇冲洗,空气干燥,以胃蛋白酶(4mg/ml,用0.2n HCl配)37℃ 30min。消化后,载片以PBS冲洗。在杂交前,载片孵育于1%甘氨酸溶液-PBS中15min,以PBS冲洗和系列酒精脱水,空气干燥。载片在80℃孵育30min使之变性。 2.杂交本实验的杂交液略区别于其它实验,否则染色体1号和18号的DNA探针不仅结合1号和18号染色体,还会同时结合染色体9,6,13,21。杂交液配方: 甲酰胺 60%(V/V,2×SSC配,pH5.0) 鲱鱼精子DNA 1μg/μl 余与一般杂交液配制相同 对多靶点ISHH,须另加入1mmol/L KCN入杂交液,每张载片加5μl的含特异性DNA探针(2.5ng/μl)的杂交液,以盖片覆盖,四周用橡皮泥封固,先在80℃热板上5~10min使DNA变性,细胞混悬液制片变性时间只需2.5min,然后在37℃杂交,孵育过夜。 3.杂交后漂洗同本节 (二)杂交后漂洗步骤。 4.显示步骤根据标记物的种类(同位素、荧光素、生物素或地高辛)分别进行显示(与第二十章 中叙述相同)。 5.结果在光镜下可见染色体结构的改变,如在膀胱肿瘤细胞,其DNA含量经FCM显示高于正常组织1倍,在双靶ISHH显示载片上,可见肿瘤细胞间期核内,1号染色体为3倍体,而18号染色体为2倍体。在肿瘤细胞内应用双重靶点或多重靶点ISHH技术可见细胞间期核内多个染色体的众多畸变相。 ISHH技术作为一项快速与敏感的侦检细胞间期细胞核内染色体畸变的新技术,可结合病理材料的常规组织切片、免疫组化染色和FCM等对病检材料做出早期的、准确的诊断,并对肿瘤的预后与进展等有所了解。本技术成功的关键在于具有特异性的DNA探针和ISHH的侦检程序,比如染色体的拷贝数与肿瘤细胞前处理的方法有关,如移除蛋白质不彻底,就会使侦检到的染色体拷贝数减少。又如在切片上进行细胞间期染色体的ISHH时,常见到一些细胞核碎片呈不同形状的斑点状(在涂片上不会出现此种图像)。对ISHH所获得资料的分析,须经过反复的实践,比如模糊的或分裂的斑点可以是非整倍体(aneuoploidy)的假阳性反应,产生于细胞周期的G2期。
四、利用性染色体特异性DNA探针的ISHH技术(一)基本原理 利用性染色体X或Y的特异性DNA探针的ISHH技术进行胎儿生前(Prenatal)性别的诊断,样品取自于羊水、绒毛或胎儿的血液。近年,科技工作者又成功地进行了胚前(pre-embry-o)或植入前(preimplantation)的ISHH诊断。所谓胚前诊断是在受精卵植入子宫内膜前的4~6细胞期,以显微操纵器(micromanipulator)或微型吸管穿破透明带,将分裂球或其中的部分细胞吸出放入培养液内,应用ISHH技术进行性染色体的分析,如有染色体畸形,可及早移除,避免以后进行人工流产术。本技术可还用于鉴别精子是带X或Y染色体。如果1个妇女遗传性基因是与X染色体相联系的,那么可给予人工授精只带X染色体的精子以避免其子代有遗传疾病的危险。当然带X染色体的精子的分离技术还有待于研究和建立。目前这还是一种实验性设想。羊水可在16~20周妊娠期吸取。在18~19周时,20ml的羊水内大约含100万个细胞,含大约7μgDNA。绒毛膜的绒毛可在3~6月取,每次可获5~50mg的组织。应注意在应用前清除母体细胞。胎血0.2~0.7ml可在17~40周采取,在15~21周,胎血中含2×109白细胞/L和类似数量的有核红细胞(Miller et al, 1985)。在32~34周,有核红细胞的比例数降至0~50/每100个白细胞。这种以性染色体为探针的ISHH技术,可应用于分裂中期的染色体铺片上,也可以应用于间期细胞核制片。性染色体ISHH技术在下列场合特别有用,如(1)当无足够的分裂细胞供可靠的细胞遗传学分析时;(2)当胎儿面临性染色体连锁遗传性疾病的危险而需做迅速的性别确定时;(3)协助鉴别45,X/46,XY镶嵌和其它异常染色体的镶嵌类型;(4)当一雌性具有一Y染色体的杂交时,应检查其父母的血样品,因为在正常情况下有部分的雄性具有2个Y染色体。这里只叙述了性染色体在ISHH技术的应用,如用同样技术标记其它染色体也可能对其它遗传性疾病进行诊断,如Julien等(1986)曾应用染色体21号DNA探针于间期细胞核,出现3倍体,为常见遗传性疾病Down氏综合征的诊断特征(发病率为1/700出生者),同样,在Edward氏综合症(遗传性疾病出现率1/3000出生者)显示18号染色体为3倍体。West实验室进行了大量的性染色体的ISHH实验,并比较了各种标记物的优缺点,他经反复实验后承认非放射性同位素标记物如生物素、地高辛具有许多优点,但在性染色体的ISHH显示,他认为至少在他的实验室同位素3H的标记优于其它的标记物。 (二)基本操作方法 1.Y染色体3H标记DNA探针原位杂交技术 (1)探针标记,以3H标记Y染色体DNA探针(详见第19章 )。 (2)原位杂交 ①RNA酶的前处理:取1ml RNA酶保存液(10mg/ml)于250ml 2×SSC内,预热于37℃水浴中。选具有适量的细胞分布的载片孵育于稀释的RNA酶溶液中,37℃,1h。系列等级乙醇脱水,70%,90%,100%乙醇各2min,空气干燥。 ②相当250ml的预杂交液(含70%甲酰胺,0.6×SSC配),70℃水浴。置载片于此预杂交液内2min以使靶DNA变性,如(1)经系列等级乙醇脱水,空气干燥。 ③DNA探针准备与变性 杂交液:10×SSC20% tRNA 10% 甲酰胺 50% 硫酸葡聚糖 20% DNA探针-3H 20% 均匀混合,70℃水浴5min使探针变性,然后迅速置冰上冷却。 10×SSC:1.2mmol/L NaCl 0.15mmol/L醋酸钠 0.2mol/L NaPO4 tRNA(SigmaR1759)4mg/ml(用TE缓冲液配),-4℃保存。 TE缓冲液:10mmol/L Tris,1mmol/L EDTA,Ph7.5。 每张载片加20μl含探针的杂交液,覆以盖玻片,四周用橡皮泥封固,于37℃孵育过夜。可测定3H的放射比性,以了解每张载片的探针含量。 ④杂交后漂洗,次日移除盖玻片,用缓冲液39℃漂洗 50%甲酰胺1×SSC 4×5min 1×SSC 39℃ 4×5min ⑤系列等级乙醇脱水,70%,90%,100%各2min,空气干燥。 (3)放射自显影(详见二十章 第一节 ):浸入核乳胶,暗盒于冷室或4℃冰箱储存,显影,定影,复染,封固和观察。应用3H标记pHY2.1DNA探针,其曝光时间大约为6~7日。 2.杂交前精子的处理 (1)取1ml用缓冲液清洗过的精子,加1ml 6mmol/L EDIT(60μl 0.2mol/L EDTA加1940μl的BWW培养基)。留置于室温5~10min,离心(1000rpm(175×g)5min。 (2)弃上清液,沉淀的精子内加1ml 2mmol/L DTT(dithiothretiol, DTT,二硫苏糖醇),配法:154μl的DTT加1846μl(4g/ml)BWW培养基(Gibbers et al, 1971),置室温45min,再离心1000rpm 5min。 (3)弃上清液,用BWW培养基再稀释、离心。 (4)加新鲜配制固定剂(甲醇:醋本以=3:1),反复混匀或置于漩涡式的混匀器上混匀,室温30~60min,离心。 (5)加数滴新鲜固定剂于离心管中,如上再混匀。迅速从混匀液中取2~3滴精液滴于载玻片上,空气干燥,以相差显微镜检查确有精子存在,保存在清洁的干盒中备用。 (6)在ISHH以前,取出载片,加数滴0.05% SDS(20μl 0.05% SDS/每2ml BWW培养基),静置5min,倾去SDS(十二烷基磺酸钠,配制法见附录2),以2×SSC漂洗,系列等级乙醇脱水,70%,90%,100%各2min,空气干燥。 (7)杂交步骤同上,杂交后以0.5%伊红黄(Eosin yellow)复染,自来水冲洗,空气干燥,DPX封固。 本节 简要介绍了ISHH技术在染色体铺片应用的三个方面,并摘要介绍了它们的应用及操作方法。由于这一技术在国际上也处于刚起步阶段,其发展主要在近10年,国内尚未开展,因此,此一技术的应用还有待于不断的摸索与完善。必须说明的是ISHH在染色体制片的应用也需设置对照实验组(详见第二十章 第一节)。
…… 第三节 DNA及寡核苷酸探针在原位杂交组织化学中的应用 第四节 原位杂交组织化学与免疫细胞化学结合法 第五节 原位杂交免疫细胞化学技术的未来与展望 参考文献 第二十一章 原位杂交技术在染色体和电镜水平的应用 第一节 原位杂交技术在染色体铺片的应用(当前页) 第二节 原位分子杂交技术在电镜水平的应用 参考文献 第二十二章 聚合酶链反应(PCR)技术的发展和应用 第一节 概述 …… |