|
《实用免疫细胞与核酸》 > 第二十二章 聚合酶链反应(PCR)技术的发展和应用
第五节 PCR各处应用模式
一、兼并引物(DegeneratePrimer)PCR密码子具有兼并性,如表22-4,单以氨基酸顺序推测编码的DNA序列是不精确的,但可以设计成对兼并引物,扩增所有编码已知顺序的核酸序列。用兼并引物时寡核苷酸中核苷酸序列可以改变,但核苷酸的数量应相同。兼并度越低,产物特异性越强,设计引物时应尽量选择兼并性小的氨基酸,并避免引物3’末端兼并,针对兼并的混合引物已成功地用于未知靶DNA的扩增、克隆和序列分析。现已成功地克隆了猪尿酸氧化酶基因、糖尿病相关肽基因和哺乳动物与禽类的嗜肝病毒基因。用脱氧肌苷(deoxyinosine;DI)引物进行PCR,可以代替编码蛋白的多种兼并密码子中的兼并碱基,DI的特异性主要受cDNA浓度影响。 表22-4 密码兼并性
注:选择使用的肽链最好避开有4~6个密码子的氨基酸
二、套式引物(NestedPrimer)PCR用第一套引物扩增15~30个循环,再用扩增DNA片段内设定的第二套引物扩增15~30个循环,这样可使待扩增序列得到高效扩增,而次级结构却很少扩增。用起始引物限量方法或Centricon30(Amicon)分子滤过器离心,在第二套引物加入前去除第一引物。此方法已成功地用来分析中国仓鼠卵巢细胞AS52的分子突变。AS52细胞含有单拷贝的细胞gpt(guanine phos-phribosytransferase)基因,与哺乳动物具有同源性。套式引物PCR减少了引物非特异性退火,从而增加了特异性扩增,提高了扩增效率。对环境样品中微生物检测和单拷贝的基因靶DNA的扩增是非常有效的。 若将套式PCR的内外引物稍加改变,延长外引物长度(至25~30bp),同进缩短内引物长度(15~17bp),使外引物先在高温退火温度下做双温循环扩增,然后改换至三温循环,使内引物在外引物扩增的基础上作低温火温度的三温循环直到扩增完成,这样就可以使两套引物一次同时加入,两种循环一气呵成,等于只做一次PCR,而灵敏度与套式二次PCR无异,在我们最近推出的PTc 51气流式DNA热循环仪上就可以完成全部程序。 套式一次PCR的成功,使PCR检测的全过程可以在5h内完成,使当天出检验报告成为现实,也使PCR检测走入临床有了现实的基础。
三、复合PCR(Multiplex PCR)用多对引物同时扩增几条DNA片段的方法称为复合PCR。这一方法最初是由Chanberlain 等检测人的基因发展而来。Bej等随之发展了对环境样品中不同属细菌相关基因序列同时PCR扩增的检测方法。两种不同的军团菌(legionella)基因,一为特异嗜肺L基因(mip),另一种为L-5SrRNA基因,通过引物摇摆(staggered)添加进行复合PCR。首先mip引物PCR扩增7个循环,然后加入5SrRNA引物PCR扩增38个循环。加入不同量的LacZ和LacB基因引物进行PCR扩增可以检测大肠杆菌和与人类粪便污染有关的细菌包括E.coli大肠菌、肠源致病沙门氏菌和志贺氏菌。 在复合PCR中,所有引物Ta值应相近。如果两对引物Tq值差异超过±℃10%,会使扩增产物的量明显不同,其中一种扩增产物或目的DNA很难观察到。另外,靶DNA的长度也应相近,差别大时短片的靶DNA会优先扩增,因此,会产生不同产量的扩增产物,为此,须采用DNA摇摆扩增或加入不等量的引物方法进行解决。
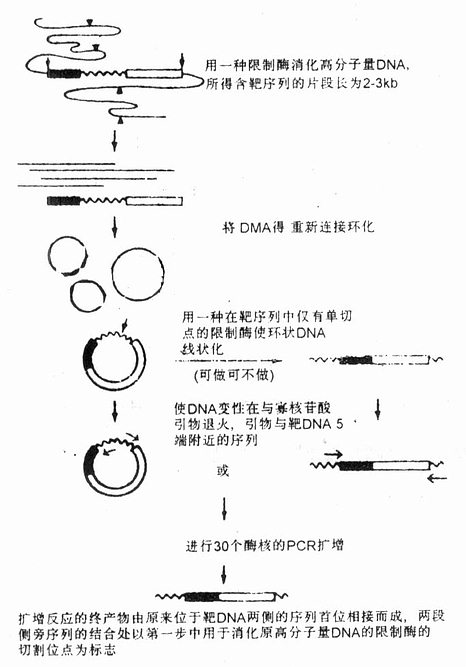
四、反向PCR(Inverse PCR或Reverse PCR)反向PCR的目的在于扩增一段已知序列旁侧的DNA,也就是说这一反应体系不是在一对引物之间而是在引物外侧合成DNA(见图22-2)。反向PCR可用于研究与已知DNA区段相连接的未知染色体序列,因此又可称为染色体缓移或染色体步移。这时选择的引物虽然与核心DNA区两末端序列互补,但两引物3’端是相互反向的。扩增前先用限制性内切酶酶切样品DNA,然后用DNA连接酶连接成一个环状DNA分子,通过反向PCR扩增引物的上游片段和下游片段;现已制备了酵母人工染色体(YAC)大的线状DNA片段的杂交探针,这对于转座子插入序列的确定和基因库染色体上DNA片段序列的识别十分重要。 该方法的不足是:①需要从许多酶中选择限制酶,或者说必须选择一种合适的酶进行酶切才能得到合理大小的DNA片段。这种选择不能在非酶切位点切断靶DNA。②大多数有核基因组含有大量中度和高度重复序列,而在YAC或Cosmid中的未知功能序列中有时也会有这些序列,这样,通过反向PCR得到的探针就有可能与多个基因序列杂交。 利用反向PCR可对未知序列扩增后进行分析,探索邻接已知DNA片段的序列,并可将仅知部分序列的全长cDNA进行分子克隆,建立全长的DNA探针。适用于基因游走、转位因子和已知序列DNA旁侧病毒整合位点分析等研究。
图22-2 反向PCR原理示意图 波浪线代表靶DNA,方块代表测翼序列,▲和△代表限制酶切位 点,寡核苷酸引物与模板互补处标有平箭头
五、不对称PCR(Asymmetric PCR)不对称PCR的基本原理是采用不等量的一对引物产生大量的单链DNA(ss-DNA)。这两种引物分别称为限制性引物与非限制性引物;其最佳比例一般为1:50~1:100,关键是限制引物的绝对量。限制性引物太多太少,均不利于制备ss-DNA。也可用普通PCR制备靶DNA双链DNA(ds-DNA),再以ds-DNA为模板,只用其中一种过量引物进行单引物PCR制备ss-DNA。 产生的ds-DNA与ss-DNA由于分子量不同可以在电泳中分开,而得到纯ss-DNA。 不对称PCR主要为测序制备ss-DNA,尤为用cD-NA经不对称PCR进行DNA序列分析是研究真核DNA外显子的好方法。
六、标记PCR(LP-PCR)和彩色PCRLP-PCR(Labelled Primers PCR)是利用同位素、荧光素等对PCR引物5’端进行标记,据此检测目的基因的存在与否,与常规PCR相比更为直观,省去了限制性内切酶酶切及分子杂交等繁琐步骤,而且一次可以同时分析多种基因成分,因而特别适合于大量临床标本的基因诊断。目前该方法只对PCR产物进行定性鉴定。 彩色PCR(Colorcomplement assay)直译为“着色互补性检测”,是LP-PCR的一种,彩色PCR意译更为明确:它用荧光染料标记引物的5’端。荧光染料JOE和FAM呈绿色荧光;TAMRA呈红色荧光;COUM呈蓝色荧光。不同荧光标记的引物同时参加反应,扩增后的目的基因会分别带有引物5’端的染料,通过电泳或离心沉淀,肉眼就可以根据不同荧光的色泽判断目标基因是否存在及扩增基因的类型。通常仅需2种不同颜色的引物,一种作为基因检测引物;另一种作为控制条件的内对照,即可诊断基因缺失、染色体易位或感染某种病毒。检测多种点突变时,可用更多的色彩,如多点突变的遗传病、几种可疑病毒感染、HLA位点分析都可以用彩色PCR同时检测多个位点。
七、加端PCR加端PCR(add-PCR)是使扩增产物的5’-末端加一段DNA顺序的PCR。设计加端PCR的引物时,除与模板配对的那一部分外再加上若干碱基,这样使扩增产物的末端加上额外一段DNA,如加上一个限制酶的识别顺序或特定功能的DNA片段。Stoflet等报道在结构基因前加上噬菌体T7的启动子,当然也可用于DNA片段的末端标记或引入特定的点突变。末端可加碱基的数量与引物的长短有关,当引物足够长时扩增产物或末端甚至可以加上十几个到几十个碱基。
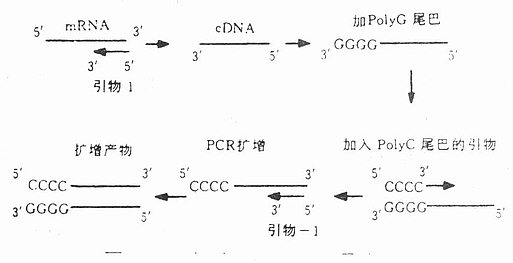
八、锚定PCR或固定PCR锚定PCR(AnchoredPCR, A-PCR)主要用于分析具有可变末端的DNA序列,Loh等用A-PCR对人外周血淋巴细胞T细胞受体α-链的mRNA的多变性进行了分析。先合成cDNA,并用末端脱氧核苷酸转移酶在其3’-可变区末端加上一个PolyG尾巴。Loh等恒定区与可变区连接部位设一个引物,另一个引物是一个具5’-polyG尾巴的引物。带有PolyG尾巴的引物是一个固定点,它可以并与PolyG尾巴结合,无论其余部分序列如何,只识别片段末端,利用此法可从前述mRNA中检出至少20种不同序列,每一种都是独特的,表明A-PCR不对任何特殊序列有倾向性结果,可用于T细胞、肿瘤及其它部位抗体基因的研究。
九、玻片PCR在聚丙烯管中可以对多种含膜板材料进行PCR,而在显微玻片上用组织细胞涂片或切片直接进行DNA扩增的方法就称为玻片PCR(Slide-PCR)。
图22-3 锚定PCR原理示意图 先将细胞涂片或呈单层细胞,然后用甲醇/醋酸(3:1,V/V)、Carnoy溶液、无水乙醇或4%多聚甲醛溶液固定5~15min。用蒸馏水冲洗,干燥,直接使用或保存于-20℃备用。在玻片上划20mm×28mm为免疫组化反应区。加入30μl PCR反应混合液,其中含10mmol/L TrispH8.3,50mmol/L KCl,1.5mmol/L MgCl2,200μmol/l dNTPs,100nmol/L引物,0.01%(V/V)明胶,0.2%BSA, 2.5u/100μl Taq酶。然后盖上22mm×40mm的盖玻片,边缘用石蜡油封好。把玻片放入PCR热循环仪金属块上,使金属块与样品呈最大程度接触,同在聚丙烯管中一样,进行30~40个循环。对于较短的扩增片段在后期循环中变性温度可降低。反应后,将致冷玻片放在氯仿中除去大部分石蜡油,但不取出盖玻片,用一个尖镊子轻轻拈起盖玻片一角,在相对的一角中PCR反应混合液呈半月形液面,用移液器回收。一般可回收25μl混合液,将反应产物进行琼脂糖电泳或用套式PCR引物按标准PCR进行重新扩增。片上扩增物可做原位杂交显示。 在Slide-PCR中,需0.1%~1%的BSA。加入BSA可以保证扩增结果,但效率不一定很高。明胶(至少0.0001%),对扩增1kb左右的靶DNA十分重要。但对小片段扩增结果影响不大。不同的样品提取方法或固定法对Slide-PCR都可行。 Silde-PCR的机理可能是在起始变性过程中一部分DNA从细胞中洗提出来,然后在细胞和玻片的水相中进行PCR。用地高辛标记的人全基因组DNA探针杂交表明在起始循环中DNA极微量,而30个循环后很丰富。常规细胞染色表明只有少量的形态改变。 Silde-PCR对于玻片上的细胞样品提供了一种较好的方法,而不必再把这些样品从玻片上括下来,使操作简便,污染减少。本方法对于原样品量极微且需病史追踪保存的(如子宫颈涂片或涂片)具有实用价值。
十、反转录PCR方法检测RNARNA的多聚酶链式反应(RT-PCR)是以RNA为模板,联合逆转录反应(reverse transcrip-tion, RT)与PCR,可用于检测单个细胞或少数细胞中少于10个拷贝的特异DNA,为RNA病毒检测提供了方便;并为获得与扩增特定的RNA互补的cDNA提供了一条极为有利和有效的途径。 RNA扩增包括两个步骤:①在单引物的介导下和逆转录酶的催化下,合成RNA的互补链cDNA;②加热后cDNA与RNA链解离,然后与另一引物退火,并由DNA聚合酶催化引物延伸生成双链靶DNA,最后扩增靶DNA。 在RT-PCR中关键步骤是RNA的逆转录,cDNA的PCR与一般PCR条件一样。由于引物的高度选择性,细胞总RNA无需进行分级分离,即可直接用于RNA的PCR。但RT-PCR对RNA制品的要求极为严格,作为模板的RNA分子必须是完整的,并且不含DNA、蛋白质和其它杂质。RNA中即使含有极微量的DNA,经扩增后也会出现非特异性扩增;蛋白质未除净,与RNA结合后会影响逆转录和PCR;残存的RNase极易将膜板RNA降解掉。硫氰酸胍(GaSCN)-CsCl法或酸性硫氰酸胍-酚-氯仿法可提得理想的RNA制品,尤以后者方法为佳,适合一般实验室进行。 常用的逆转录酶有两种,即禽类成髓细胞性白血病病毒(Avianmyeloblastosis virus, AMV)和莫洛尼鼠类白血病病毒(Moloney murineleukemia virus, MO-MLV)的逆转录酶(RT)。一般情况下用Mo-MLV-RT较多,但模板RNA的二级结构严重影响逆转录时,可改用AMV-RT,因后者最适温度为72℃,高于Mo-MLV-RT的最适温度(37℃),而较高的反应温度有助于消除RNA的二级结构。 一步法扩增(one step amplification)是为了检测低丰度mRNA的表达,利用同一种缓冲液,在同一体系中加入逆转录酶、引物、Taq酶、4种dNTP直接进行mRNA反转录与PCR扩增。发现Taq酶不仅具有DNA多聚酶的作用,而且具有反转录酶活性,可利用其双重作用在同一体系中直接以mRNA为模板进行反转录和其后的PCR扩增,从而使mRNA的PCR步骤更为简化,所需样品量减少到最低限度,临床小样品的检测非常有利。用一步法扩增可检测出总RNA中小于1ng的低丰度mRNA。该法还可用于低丰度mRNA的cDNA文库的构建及特异cD-NA的克隆,并有可能与Taq酶的测序技术相组合,使得自动反转录、基因扩增与基因转录产物的测序在一个试管中进行。 最近Cetus公司推出rTth逆转录酶,兼具有DNA多聚酶的活性,对于RNA的发展起了很大的推动作用。有关rTth逆转录酶法参考第四节 有关内容。
十一、定量PCR1.DNA-PCR定量用同位素标记的探针与电泳分离后的PCR扩增产物进行杂交,根据放射自显影后底片曝光强弱可以对模板DNA进行定量。Abbot等利用这种方法对人类T细胞白血病反转录基因进行了定量研究。 PCR扩增产物用专为检测ds-DNA而设计的微量荧光计定量,利用染料H33258专与双链DNA结合而使荧光增强50倍的特性。可以从标准模板系列稀释扩增产物量曲线上读出样品中模板DNA的量或拷贝数,达到PCR定量的目的。 利用倍比稀释模板作系列稀释PCR,求出最低(PCR-EB)检测限来比较,也是常用的半定量PCR方法。 2.mRNA-PCR定量由于MRNA-PCR定量需经两个酶(RT和Taq)催化,因而影响因素较多。1989年Wang等报道了低丰度mRNA绝对定量方法。利用浓度已知且与待测靶mR-NA序列相同的内对照mRNA(其片段长短不同,便于PCR扩增后产物的分离),在同一体系中,用相同的由32P标记的引物与待测mRNA一同进行逆转录和PCR扩增,扩增产物电泳后,分别测定二者产物放射性强度,由预先制备的标准曲线推算出每个样本特异mRNA的量。Gilliland等的结果表明,在1ng总RNA中可以对小于1pg的特异mRNA进行定量。这一定量方法在肿瘤、代谢失调、基因表达调控等研究中均有重要意义。
…… 参考文献 第二十二章 聚合酶链反应(PCR)技术的发展和应用 第一节 概述 第二节 PCR技术的原理 第三节 PCR操作范例及反应体系的组成 第五节 PCR各处应用模式(当前页) 第六节 样品处理与注意事项 第七节 PCR仪器的发展 第八节 PCR临床应用领域及特点 第九节 其它链扩增技术及应用范围 …… |