|
《药理学》 > 第二章 药物效应动力学
第四节 药物与受体受体(receptor)是细胞在进化过程中形成的细胞蛋白组分,能识别周围环境中某种微量化学物质,首先与之结合,并通过中介的信息转导与放大系统,触发随后的生理反应或药理效应。自从Langley 提出受体学说100年后,受体已被证实为客观存在的实体,类型繁多,作用机制多已被阐明,现在受体已不再是一个空泛笼统的概念。受体分子在细胞中含量极微,1mg 组织一般只含10fmol左右。能与受体特异性结合的物质称为配体(ligand)。受体仅是一个“感觉器”,对相应配体有极高的识别能力。受体-配体是生命活动中的一种偶合,受体都有其内源性配体,如神经递质、激素、自身活性物(autocoid)等。能激活受体的配体称为激动药(agonist),能阻断其活性的配体称为拮抗药(antagonist)。根据受体与配体结合的高度特异性,受体被分为若干亚型,如肾上腺素受体又分为α1、α2、β1和β2等亚型,其分布及功能都有区别。受体与配体有高度亲和力,多数配体在1pmol~1nmol/L的浓度时即可引起细胞的药理效应。反应之所以如此灵敏主要是靠后续的信息转导系统,如细胞内第二信使(secondmessenger)的放大、分化及整合功能。酶、载体、离子通道及核酸也可与药物直接作用,但这些物质本身具有效应力,故严格地说不应被认为是受体。某些细胞蛋白组分可与配体结合,但没有触发效应的能力,称为结合体(acceptor)。
一、受体动力学受体动力学一般用放射性同位素标记的配体(L)与受体(R)做结合试验研究。取一定量组织,磨成细胞匀浆,分组加入不同浓度的放射性同位素标记的配体(药物),温孵待反应达平衡后,迅速过滤或离心分出细胞,用缓冲液洗去尚未结合的放射性配体,测定标本的放射强度,这是药物与细胞结合的总量,此后用过量冷配体(未用同位素标记的配体)洗脱特异性与受体结合的放射性配体再测放射强度,这是药物非特性结合量。将总结合量减去非特性结合量就可以获得L-R结合(B)曲线。如果L只与单一R可逆性结合,以B为纵座标,[L]为横座标,L-R结合曲线为直方双曲线(图2-5)。如将横座标改用log[L]([]表示摩尔浓度)则呈典型的S形量效曲线。 按质量作用定律
(E代表效应) 反应达到平衡时
(KD是解离常数) 因为[RT]=[R]+[LR](RT为受体总量),代入上式并经推导得
由于只有LR才发挥效应,故效应的相对强弱与LR相对结合量成比例,即
按此公式以E为纵座标,log[L]为横座标作图,结果与实验数据图形完全一致。
当[L]=0时,效应为0, 当[L]>>KD时,[LR]/[RT]=100%,达最大效能,即[LR]max=[RT]。 当[LR]/[RT]=50%时,即EC50时,KD=[L]。 KD表示L与R的亲和力(affinity),单位为摩尔。各药(L)与R亲和力不同,KD越大时亲和力越小,二者成反比。令pD2=-logKD则其值不必用摩尔单位、数值变小且与亲和力成正比,在半对数座标上也较易理解,故pD2较为常用。 药物与受体结合产生效应不仅要有亲和力,还要有内在活性(intrinsicactivity),后者用α表示,0≤α≤100%。故上述公式应加入这一参数:E/Emax=α[LR]/[RT]。两药亲和力相等时其效应强度取决于内在活性强弱,当内在活性相等时则取决于亲和力大小(图2-6)。 将上述受体动力学基本公式([LR]/[RT]=[L]/KD+[L])加以推导改变可将S形量效曲线改变为直线关系,使计算方便很多也准确很多: 1.双倒数图 将上述基本公式两侧取倒数后加以推导得1/[LR]=KD/[L][RT]+1/[RT]。以1/[LR]为纵座标、1/[L]为横座标作图得直线(图2-7),斜率为KD[RT],即KD/Emax,与纵座标交点为1/[RT],即1/Emax,与横座标交点为-1/KD。 2.Scatchard图 推导得公式[LR]/[L]=[RT]/KD-[LR]/KD以[LR]/[L],为纵座标,[LR]为横座标作图也呈直线(图2-8),斜率为-1/[KD],与纵座标交点为[RT]/KD,与横座标交点为[RT]。 这些直线关系图解在受体研究中有重要用途,也可加深对受体动力学的理解
图2-6 药物与受体的亲和力及其内在活性对量效曲线的影响 A图 a,b,c三药与受体的亲和力(pD2)相等,但内在活性(Emax)不等 B图 a,b,c 三药与受体的亲和力(pD2)不等,但内在活性(Emax)相等
图2-7 受体结合量效关系的双倒数作图
图2-8 受体结合量效关系的Scatchard作图 一些活性高的药物与相应受体结合的量效曲线 (B-log[L]曲线)并不一定与结合后产生效应的量效曲线(E-log[L]曲线)相重合。因为这类药物只需与一部分受体结合就能发挥最大效应(Emax),剩余下未结合的受体为储备受体(spare receptor)。这对理解拮抗药作用机制有重要意义,因为这类拮抗药必须在完全占领储备受体后才能发挥其拮抗效应。 受体激动药(L)对相应受体有较强的亲和力,也有较强的内在活性,α达100%。受体拮抗药(I)虽然也有较强的亲和力,但缺乏内在活性,α=0,本身不能引起效应,却占据一定量受体,拮抗激动药的作用。竞争性拮抗药(competitiveantagonist)能与激动药互相竞争与受体结合,这种结合是可逆性的。在实验中如果L与I同时存在则[RT]=[R]+[LR]+[IR],代入上述基本公式并加推导得
可见L和I同时存在时,如L这一因素固定不变,药理效应大小取决于[I]/K1(K1是I的解离常数)。[I]越高及(或)K1越小时效应越弱,即拮抗效果越强。当[L]>>[I]时,[LR]/[RT]→100%,这就是竞争性拮抗药使量效曲线平行右移(Emax不变)的理论解释(图2-9)。 在有一定量的竞争性拮抗药[I]存在时,增加[L]至[L’]仍可使药理效应维持在原来单用[L]时的水平。据此,
将之推导得
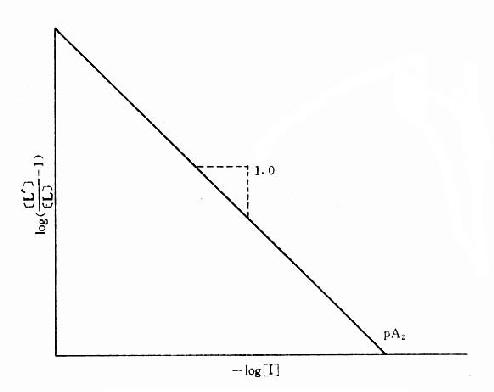
[L’]/[L]是剂量比 (dose ratio),即将[L]增加[L’]/[L]倍就能克服[I]的拮抗作用。该比值也取决于[I]/K1而与[L]绝对值或KD无关。将此公式两侧取log,并以log([L’]/[L]-1)为纵座标、以-log[I]为横座标作图,呈直线,斜率为1,与横座标交点为-logK1,即pA2此即Schild 图(图2-10)。按Schild定义,拮抗参数pAx是指剂量比为X时竞争性拮抗药浓度的负对数值。常用pA2,即[L’]/[L]=2时的数值,则pA2=-log[I]=-logK1,些参数反映拮抗药的拮抗强度,其值越大表示拮抗作用越强。
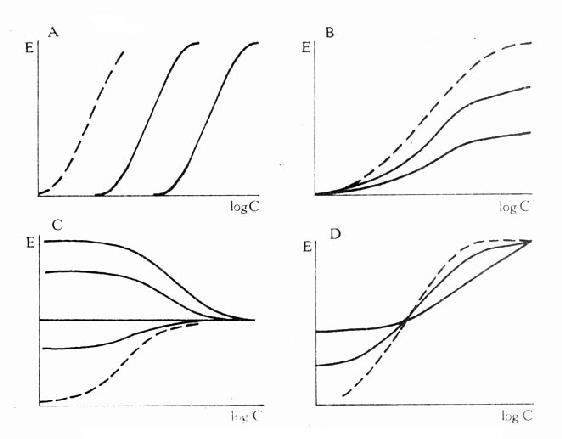
图2-9 竞争性拮抗药(A图)、非竞争性拮抗药(B图)及部分 激动药(D图)对激动药(虚线)量效的影响及激动药(C图) 对部分激动药(虚线)量效曲线的影响
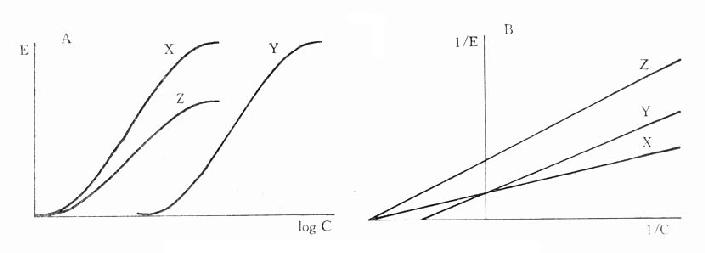
图2-10 竞争性拮抗作用的Schild作图 非竞争性拮抗药(noncompetitiveantagonist)与R结合非常牢固,分解很慢或是不可逆转,使能与L结合的R数量减少。另一类非竞争性拮抗药可阻断受体后某一中介反应环节而使受体-效应功能容量减少。二者共同特点是使量效曲线高度(Emax)下降。但L与剩余的R结合动力学不变,即KD不变。在双倒数图中更易看出这一关系(图2-11)。
图2-11 竞争性拮抗作用与非竞争性拮抗作用比较 A图 量效曲线 B图 双倒数曲线 X 单用激动药 Y 竞争性拮抗药对激动药的拮抗作用 Z 非竞争性拮抗药对激动药的拮抗作用 还有一类药物称为部分激动药(partialagonist)和R结合的亲和力不小,但内在活性有限,α<100%,量效曲线高度(Emax)较低。与激动药同时存在时,当其浓度尚未达到Emax时,其效应与激动药协同,超过此限时则因与激动药竞争R而呈拮抗关系,此时激动药必需增大浓度方可达到其最大效能。可见部分激动药具有激动药与拮抗药两重特性。(图2-9C、D) 目前放射性配体-受体结合技术已普遍用于受体研究,但必需和药理效应实验结合进行才有意义。 为什么化学结构类似的药物作用于同一受体有的是激动药,有的是拮抗药,还有的是部分拮抗药?还可用二态模型(two-statemodel) 学说解释。按此学说,受体蛋白有两种可以互变的构型状态:静息状态(R)与活动状态(R*)(图2-12)。静息时平衡趋向R。活动药只与R*有较大亲和力,L-R*结合后充分发挥药理效应。部分激动药(P)与R及R*都能结合但对R*的亲和力大于对R的亲和力,故只有部分受体被激活而发挥较小的药理效应。拮抗药对R及R*亲和力相等,且能牢固结合,但保持静息状态时两种受体状态平衡,拮抗药不能激活受体但能阻断激动药作用。个别药物(如苯二氮
二、受体类型根据受体蛋白结构、信息传导过程、效应性质、受体位置等特点,受体大致可分为下列4类: 1. 含离子通道的受体 又称直接配体门控通道型受体,它们存在于快速反应细胞的膜上,由单一肽链反复4次穿透细胞膜形成1个亚单位,并由4~5个亚单位组成穿透细胞膜的离子通道,受体激动时离子通道开放使细胞膜去极化或超极化,引起兴奋或抑制效应。最早发现的N型乙酰胆碱受体就是由α×2、β、γ、δ5个亚单位组成的钠离子通道,在α亚单位上各有一个乙酰胆碱结合点(图2-13A)与乙酰胆碱结合后,钠离子通道开放,胞外钠离子内流、细胞膜去极化、肌肉收缩。这一过程在若干毫秒内完成(钠离子通道开放时间仅1ms)。脑中γ氨基丁酸(GABA)受体情况类似,其他如甘氨酸、谷氨酸、天门冬氨酸受体都属于这一类型。
2.G-蛋白偶联受体 这一类受体最多,数十种神经递质及激素的受体需要G-蛋白介导其细胞作用,例如肾上腺素、多巴胺、5-羟色胺、M-乙酰胆碱、阿片类、嘌呤类、前列腺素及一些多肽激素等的受体,这些受体结构非常相似,都为单一肽链形成7个α-螺旋来回穿透细胞膜,N-端在细胞外,C-端在细胞内,这两段肽链氨基酸组成在各种受体差异很大,与其识别配体及转导信息各不相同有关。胞内部分有G-蛋白结合区(图2-13B)。G-蛋白(G-protein)是鸟苷酸结合调节蛋白的简称,存在于细胞膜内侧,由三个亚单位组成。主要有两类,其一为兴奋性G-蛋白(GS),霍乱弧菌毒素能使之活化,激活腺苷酸环化酶(AC);另一为抑制性G-蛋白(Gi),抑制AC,百日咳杆菌素抑制之。G-蛋白还介导心钠素及NO对鸟苷酸环化酶(GC)的激活作用。此外G-蛋白对磷脂酶C、磷脂酶A2、Ca2+、K+离子通道等有重要调节作用。一个受体可激活多个G-蛋白,一个G-蛋白可以转导多个信息给效应机制,调节许多细胞功能。 3.具有酪氨酸激酶活性的受体 这一类细胞膜上的受体由三个部分组成(图2-13C),细胞外有一段与配体结合区,中段穿透细胞膜,胞内区段有酪氨酸激酶活性,能促其本身酪氨酸残基的自我磷酸化而增强此酶活性,再对细胞内其他底物作用,促进其酪氨酸磷酸化,激活胞内蛋白激酶,增加DNA及RNA合成,加速蛋白合成,从而产生细胞生长分化等效应。胰岛素、胰岛素样生长因子、上皮生长因子、血小板生长因子及某些淋巴因子(lymphokines)的受体属于这一类型。 4.细胞内受体 甾体激素受体存在于细胞浆内,与相应甾体结合后分出一个磷酸化蛋白,暴露与DNA结合区段,进入细胞核能识别特异DNA碱基区段并与之结合促进其转录及以后的某种活性蛋白增生(图2-13D)。甲状腺素受体存在于细胞核内,功能大致相同。这两种受体触发的细胞效应很慢。需若干小时。 A.直接配体门控通道型
图2-13 受体类型示意图
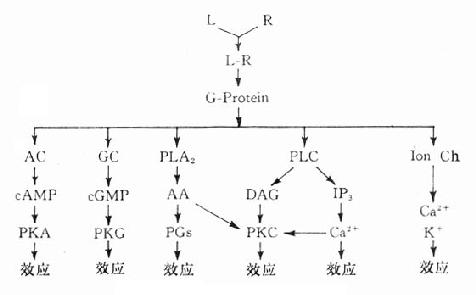
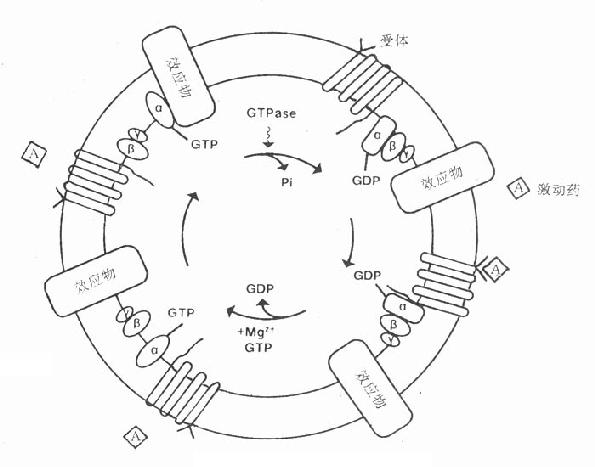
三、第二信使受体在识别相应配体并与之结合后需要细胞内第二信使(secondmessenger) 将获得信息增强、分化、整合并传递给效应机制才能发挥其特定的生理功能或药理效应。最早发现的第二信使是环磷腺苷(cAMP),现在知道还有许多其他物质参与细胞内信息转导。这是一个非常复杂的系统,简示如下(图2-14),很多问题尚有待进一步阐明。 1.G-蛋白 G蛋白是一类存在于细胞膜内侧的调节蛋白,都是由三个不同亚单位α、β、γ组成的三聚体。静息状态时与GDP结合。相应受体激活后GDP-α、β、γ复合物在Mg2+参与下,结合的GDP与胞浆中GTP交换,GTP-α与β、γ分离并与相应的效应机制结合,同时配体与受体分离。α亚单位内在的GTP酶活性促使GTP水解为GDP,激活效应机制,从而恢复原来静息状态(图2-15)。GS激活腺苷酸环化酶(AC),使cAMP增加。Gi抑制AC,使cAMP减少,G-蛋白还激活磷脂酶C(PLC),调节Ca2+、K+等离子通道。对鸟苷酸环化酶也有激活作用,作用非常广泛,介导多种效应。近来发现G-蛋白还介导激活磷脂酶A2(PLA2)而产生花生四烯酸(AA),后者是各种前列腺素及白三烯的前体。
图2-14 第二信使系统示意图 2. 环磷腺苷(cAMP) cAMP是ATP经AC作用的产物。β受体、D1受体、H2受体等激动药通过GS作用使AC活化,ATP水解而使细胞内cAMP增加。α受体、D2受体、MACh受体、阿片受体等激动药通过Gi作用抑制AC,细胞内cAMP减少。cAMP受磷酸二酯酶(phosphodiesterase,PDE)水解为5’AMP后灭活。茶碱抑制PDE而使胞内cAMP增多。cAMP能激活蛋白激酶a (PKA)而使胞内许多蛋白酶磷酸化(ATP提供磷酸基)而活化,例如磷酸化酶、脂酶、糖原合成酶等活化而产生能量。钙离子通道磷酸化后激活,钙离子内流而使神经、心肌、平滑肌等兴奋。
图2-15 G-蛋白作用示意图 3.环磷鸟苷(cGMP) cGMP是GTP经鸟苷酸环化酶(GC)作用的产物,也受PDE灭活。cGMP作用与cAMP相反,使心脏抑制、血管舒张、肠腺分泌等。CGMP可以独立作用而不受cGMP制约。cGMP可激活蛋白酶G而引起各种效应。 4.肌醇磷脂(phosphatidylinositol) 细胞膜肌醇磷脂的水解是另一类重要的受体信息转导系统。α、H1、5-HT2、M1、M3等受体激动药与其受体结合后通过G-蛋白介导激活磷脂酶C(PLC)PLC使4,5-二磷酸肌醇磷脂(PIP2)水解为二酰甘油(DAG)及1,4,5-三磷酸肌醇(IP3)。DAG在细胞膜上激活蛋白激酶C(PKC),使许多靶蛋白磷酸化而产生效应,如腺体分泌,血小板聚集,中性粒细胞活化及细胞生长、代谢、分化等效应。IP3能促进细胞内钙池释放Ca2+,也有重要的生理意义。 5.钙离子 细胞内Ca2+浓度在1μmol/l以下,不到血浆Ca2+的0.1%,对细胞功能有着重要的调节作用,如肌肉收缩、腺体分泌、白细胞及血小板活化等。细胞内Ca2+可从细胞外经细胞膜上的钙离子通道流入,也可从细胞内肌浆网等钙池释放,两种途径互相促进。前者受膜电位、受体、G-蛋白,蛋白激酶A(PKA)等调控,后者受IP3作用而释放。细胞内Ca2+激活蛋白激酶C(PKC),与DAG有协同作用,共同促进其他信息传递蛋白及效应蛋白活化。很多药物通过对细胞内Ca2+影响而发挥其药理效应,故对细胞内Ca2+调控及其作用机制近年来受到极大的重视。
四、受体的调节受体虽是遗传获得的固有蛋白,但并不是固定不变的,而经常代谢转换处于动态平衡状态,其数量,亲和力及效应力经常受到各种生理及药理因素的影响。连续用药后药效递减是常见的现象,一般称为耐受性(tolerance)、不应性(refractoriness)、快速耐受性(tachyphylaxis)等。由于受体原因而产生的耐受性称为受体脱敏(receptordesensitization)。N2-ACh受体在受激动药连续作用后若干秒内发生脱敏现象,这是由于受体蛋白构象改变,钠离子通道不再开放所致。β-Adr受体脱敏时不能激活AC是因为受体与G-蛋白亲和力降低,或由于cAMP上升后引起PDE负反馈增加所致。具有酪氨酸激酶活性的受体可被细胞内吞(endocytosis)而数目减少,这一现象称为受体数目的向下调节(down regulation)。受体与不可逆拮抗药结合后其后果等于失去一部分受体,如银环蛇咬伤中毒时,N2-ACh受对激动药脱敏。与此相反,在连续应用拮抗药后受体会向上调节(up regulation),反应敏化。例如长期应用β-Adr受体拮抗药后,由于受体向上调节,突然停药时会出现反跳反应。
…… 绪言 第二章 药物效应动力学 第一节 药物的基本作用 第二节 药物剂量与效应关系 第三节 药物作用机制 第四节 药物与受体(当前页) 第三章 药物代谢动力学 第一节 药物体内过程 第二节 体内药量变化的时间过程 第三节 药物消除动力学 …… |
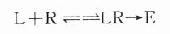

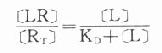





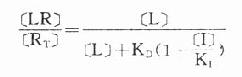
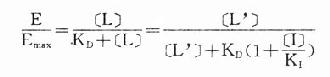

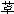 类)对R亲和力大于R*,结合后引起与激动药相反的效应,称为超拮抗药(superantagonist)。这一学说容易理解,但有待进一步实验证实。
类)对R亲和力大于R*,结合后引起与激动药相反的效应,称为超拮抗药(superantagonist)。这一学说容易理解,但有待进一步实验证实。